关于骨科脊柱产品申报美国上市前通告和国内注册申报资料的要求对比研究
孔玮娜,程云章
上海理工大学 医疗器械与食品学院,上海 200093
[摘 要]新修订的《医疗器械注册管理办法》(食品药品监督管理总局令第4号)及配套的《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告》(食品药品监督管理总局令第43号)引入了美国FDA 申报510k的思路,本文通过将《医疗器械注册申报资料要求及说明》和美国FDA《传统和简化510k的格式指导原则》(Guidance for Industry and FDA Staff Format for Traditional and Abbreviated 510ks)做对比,以骨科脊柱产品为例,浅析二者之间的异同点,以期对医疗器械注册申报资料的准备工作提供指导和帮助。
[关键词]医疗器械;注册;骨科脊柱产品
医疗器械注册申报资料是医疗器械安全有效上市的证明文件,文件的质量高低决定了产品能否通过评审,以及能否走向市场。而科学细化的注册申报资料要求,能够给予企业明确的申报思路,缩短上市时间。通过对比美国上市前通告(简称510k)申报资料与我国新修订的注册申报资料要求发现,新要求不仅结合了欧盟和美国等发达国家的经验,且有较大区别,主要区别梳理总结如下。
1 申报资料框架对比
新要求对于申报资料要求有明确的目录清单,包含一级标题和二级标题,有些文件如医疗器械安全有效基本要求、产品技术要求、临床评价资料等,企业可按照目录逐条准备文件,不用另外设计格式。相同的目录格式可减少审评人员的审评时间和工作量。我国医疗器械注册申报资料和美国510k申报资料框架对比,见表1。
表1 申报资料框架对比


从框架上看,新注册资料要求中,第1部分申请表,与510k的第2部分CDRH上市前审核提交的表格类似,规定了所属型号、申报者、联系人地址、联系方式以及器械分类等信息。510k的要求更为详细,其第3部分还包括了封面信件,对比其第2部分,提供了更详细的申报信息。
新注册资料第3部分医疗器械安全有效基本要求清单参考了欧盟的要求,这点在510k中没有要求。第4部分综述资料,与510k中第11部分器械描述类似,要求描述产品的工作原理、机理组成、结构组成(配合使用的配件)、各规格型号的区别。但是我国的注册资料要求这部分还需要提供包装说明、适用范围、禁忌症、参考的同类产品和前代产品的信息、研发背景和目的。目的是使审评员在不看其他文件的情况下,可以完整地了解产品概况。
第5部分研究资料是与510k最为相似的文件,它要求提供产品性能研究,如生物学评价、软件研究、电气安全、电磁辐射和临床研究等内容,这与510k中第14~20部分相似。但是510k中未要求提供生产制造信息、临床评价资料、产品风险分析资料、产品技术要求和注册检测报告,只要求企业做相应的测试研究,并提供测试报告。而我国的注册申报资料要求比510k更为严格,需要制定技术要求并提供给有资质的检测机构做注册检验,取得注册报告后才能申报注册。而且还需要提供生产加工工艺,注明关键工艺和特殊工艺,以及过程控制点。明确生产过程中各种加工助剂的使用情况及对杂质控制的情况。
510k中的第12部分实质性等同讨论是申报资料的核心,是指通过对拟上市产品与已上市产品在安全性和有效性方面进行比较,得出“实质性等同”的结论。我国的新注册申报资料第7部分临床评价资料要求提出“同品种医疗器械”的概念,在过去这些产品是需要做临床试验的,但是通过同品种产品的对比,如果支持性文件充分,企业也可以申请免临床试验。这点是我国法规向国际化接轨的一个重要标志。
2 骨科椎弓根螺钉系统的申报要求区别
美国21CFR 第888部分骨科器械的规定,椎弓根螺钉系统根据不同的适用范围,属于美国Ⅱ、Ⅲ类产品,由于Ⅲ类产品还没有明确的上市前批准(PMA)要求,目前按照510k的途径申报。所以本文只讨论510k的申报要求。而中国将此类产品归为Ⅲ类医疗器械,需要国家食品药品监督管理总局(CFDA)审批。
美国在2004年5月3日发布了《脊柱系统申报510k的指导原则》(Guidance for Industry and FDA Staff Spinal System 510ks),与国家食品药品监督管理局医疗器械审评中心发布的《无源植入性医疗器械注册申报资料指导原则》类似,只是后者尚未根据新法规进行更新。前者对于具体的一类产品提出了较为详细建议,例如机械性能测试项目、动物研究、临床研究、灭菌要求、标签和说明书等。两者对比主要有以下2点区别:
(1)我国还没有发布针对脊柱系统的注册申报要求,只对无源植入性医疗器械的注册申报有总的要求,且后者是按照老法规的规定提出的,尚未更新。
(2)前者对于产品描述需要提供的信息要求比较有操作性,例如:① 包含产品组件名称、产品代号、尺寸等信息的表格示例;② 完整的包含尺寸的工程图纸;③ 各组件之间的连接方式,可以提供图片等信息;④ 脊柱模型的放大照片或草图;⑤ 使用到的手术器械的信息(如手术器械清单、图纸、材料、符合的材料标准)不同的脊柱系统,如用于颈前路系统、胸腰椎后路系统、前外侧和胸腰椎后路系统等各系统的适应症并且需要有举例说明。而后者对于综述资料则要求提供:① 产品分类与产品命名、型号、规格;② 产品作用机理,预期与人体接触部位、接触方式、作用时间,预期与人体接触时间的确定依据及相关研究资料、主要原材料;③ 产品性能、结构及组成。相比较前者,提供示范会给企业一个明确的指导。
对于机械性能测试部分,前者对于颈椎系统、非颈椎系统推荐的测试项目为:静态和动态轴向压缩弯曲试验、静态扭转试验。而在我国2015年7月1日实施了一系列脊柱植入物推荐性行业标准,其与ASTM标准的关系,见表2。YY/T0119.1~5-2014《脊柱植入物-脊柱内固定系统》系列标准,强调了单个部件-脊柱内固定系统部件的要求。
表2 行业标准与美国ASTM标准的关系
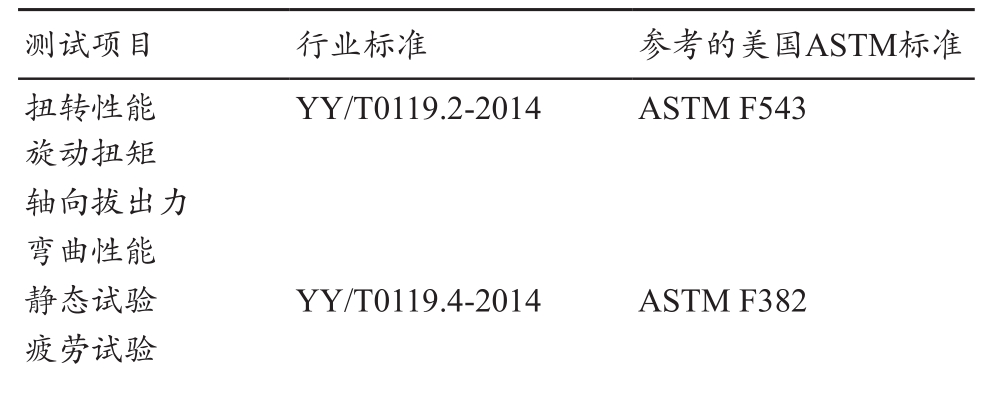

YY/T0961-2014《脊柱植入物 脊柱内固定系统组件及连接装置的静态及疲劳性能评价方法》,规定了脊柱内固定系统单轴静态和疲劳强度以及组件连接装置抗松动性的试验方法,旨在为不同设计的脊柱植入物连接装置提供力学性能评价方法[5-6]。
YY/T0857-2011《椎体切除模型中脊柱植入物试验方法》反映了脊柱植入物的最高水平,用于评价由多个部件装配而成的完整系统,这种系统涉及许多组件及其相互连接的作用。
上述标准虽然在国内已经实施,但由于是推荐性标准,目前尚未明确企业注册时是否需要在指定的医疗器械检测中心做所有推荐性检测项目的注册检验或者只做整套系统的测试,如YY/T0857。
3 对我国骨科脊柱系统注册申报资料要求的建议
通过将我国的《医疗器械注册申报资料要求及说明》与美国510k申报资料要求对比,以及《脊柱系统申报510k的指导原则》与《无源植入性医疗器械注册申报资料指导原则》的对比,有以下3点建议:① 医疗器械审评中心加快修订或制定骨科脊柱产品、关节产品等申报资料指导原则,提供示范给企业做参考,这样可以提高申报资料的质量,统一尺度,加快审评速度;② 加强事后监管,技术审核时提供研发相关的资料,一些资料如风险分析资料可以在GMP审查、飞行检查时,由企业提供。因为风险分析资料作为质量体系文件中的一部分,而且是动态的,完全可以在后续的审核中进一步检查;③ 由于机械测试项目周期长,发补后只有1年的时间补充资料,如果企业不明确审评的思路,缺少部分检测报告,可能会导致在有限的时间内无法完善测试报告,所以建议明确指出推荐性标准及需要递交的注册检验测试项目。
综上所述,我国的注册申报资料要求已经从国外获取了经验,学习了其中的先进之处,完善了注册文件的要求。但是对于某些类型的医疗器械,企业申报中还存在一些疑问,还应该继续明确提出标准要求,逐步填补漏洞,不断完善注册申报资料的要求。
[参考文献]
[1] 国家食品药品监督管理总局.关于公布医疗器械注册申报资料要求和批准证明文件格式的公告(2014年第43号)[EB/ OL].2014-09-05.
[2] 国家食品药品监督管理总局.无源植入性医疗器械注册申报资料指导原则[EB/OL].2009-12-30.
[3] CDRH. Guidance for Industry and FDA Staff Format for Traditional and Abbreviated 510(k)s[EB/OL].http://www.fda.gov/ MedicalDevices/DeviceRegulationandGuidance/,2005-08-12.
[4] CDRH.Guidance for Industry and FDA Staff Spinal System 510(k)s[EB/OL].http://www.fda.gov/MedicalDevices/DeviceR egulationandGuidance/,2004-05-03.
[5] 王小健,苏云星,常峰,等.脊柱手术前椎体定位装置的研制及临床应用[J].中华实验外科杂志,2015,32(7):1758.
[6] 何剑颖,董谢平,舒勇,等.脊柱保护器对腰椎保护的有限元分析[J].中国矫形外科杂志,2015,23(6):548-555.
A Comparative Study on the Premarket Notification in the United States and the Requirements for Domestic Registration for Orthopedic Spinal Products
KONG Wei-na, CHENG Yun-zhang
School of Medical Instrument and Food Engineering, University of Shanghai for Science and Technology, Shanghai 200093, China
Abstract:The newly revised Provisions forMedical Device Registration (CFDA No.4)andRequirements and Instructions for Medical Device Registration Application (CFDA No.43)introduced the idea of premarket notification 510k of US Food and Drug Administration (FDA). This study made a comparison betweenthe Requirements and Instructions for Medical Device Registration Applicationand theGuidance for Industry with the Guidance for Industry and FDA Staff Format for Traditional and Abbreviated 510ks. By using the orthopedic spinal products as examples, this paper analyzes the similarities and differences between the two documents, in order to provide guidance and assistance for the medical device registration dossiers preparation.
[中图分类号]R197.39
[文献标志码]A
doi:10.3969/j.issn.1674-1633.2016.02.056
[文章编号]1674-1633(2016)02-0172-03
收稿日期:2015-09-10
Abstract:: medical devices; registration; orthopedic spinal products